The availability of a growing number of completely sequenced genomes opens new opportunities for understanding of complex biological systems. Success of genome-based biology will, to a large extent, depend on the development of new approaches and tools for efficient comparative analysis of the genomes and their organization. We have developed a technique for detecting possible functional coupling between genes based on detection of potential operons. The approach involves computation of "pairs of close bi-directional best hits", which are pairs of genes that apparently occur within operons in multiple genomes. Using these pairs, one can compose evidence (based on the number of distinct genomes and the phylogenetic distance between the orthologous pairs) that a pair of genes is potentially functionally coupled. The technique has revealed a surprisingly rich and apparently accurate set of functionally coupled genes. The approach depends on the use of a relatively large number of genomes, and the amount of detected coupling grows dramatically as the number of genomes increases.
Key words: microbial genomes; operons; gene function identification; genetic sequence analysis; comparative analysis
The availability of a growing number of the completely sequenced genomes
opens new opportunities. Success of genome-based biology [1] will to a
large extent depend on the development of new approaches and tools for
efficient comparative analysis of the genomes and their organization.
The issue of how to determine the function of some specific gene within
a genome has been present for many years.
The desire to rapidly characterize large
numbers of genes from the newly available genomes has led to a growing
sense of urgency, as well as a number of ambitious experimental projects
to address the need.
We have developed an algorithm to detect potential operons based on
comparative of multiple genomes.
It depends on the availability of a relatively large number of genomes,
however, it does not require these genomes be complete.
Furthermore, it provides significant clues as to the function
of many genes of unknown or hypothetical function.
Our initial results indicate that at least a fifth of the sequenced genes
show detectable functional coupling by this method,
and that there is every reason to expect this percentage to grow rapidly
as the number of sequenced genomes increases.
It is well known that in many organisms the genes responsible for related functions are located close to each other on the chromosome. If one could accurately predict operons, the availability of a growing number of prokaryotic genomes would offer numerous significant clues relating to the function of "hypothetical proteins" (those for which no reliable identification of function has yet been made). However, an accurate prediction of operons is nontrivial, and the only reliable identifications are currently based on wet-lab experimentation. Nevertheless, given the growing number of genomes, it is reasonable to ask whether accurate predictions of potential operons could be based on a weaker (but related) notion of functional coupling. In this paper, we propose a straightforward computation that is remarkably accurate in predicting functional coupling of genes.
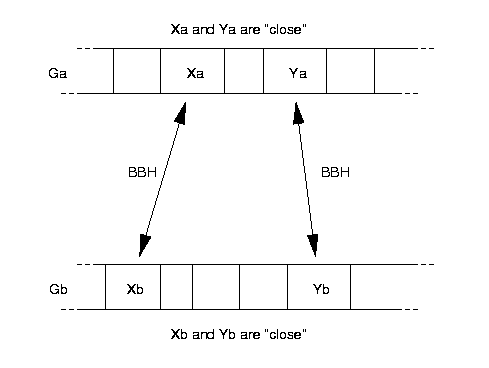
We begin with a small set of definitions that will be used throughout the rest of this paper:
A set of genes occurring on a prokaryotic chromosome will be called a "run" if and only if they all occur on the same strand, and the gaps between adjacent genes are 300 base pairs or less. Any pair of genes from within a single run are called "close".
If we have two genes Xa and Xb from two genomes Ga and Gb, Xa and Xb are called "a bidirectional best hit (BBH)" if and only if
- recognizable similarity exists between Xa and Xb (in our case, we required FASTA3 scores better than 1.0e-5),
- there is no gene Yb in Gb that is more similar to Xa than Xb is, and
- there is no gene Ya in Ga that is more similar to Xb than Xa is.
Genes (Xa,Ya) from Ga and (Xb,Yb) from Gb form a "pair of close bidirectional best hits (PCBBH)" if and only if
- Xa and Ya are close,
- Xb and Yb are close,
- Xa and Xb are a BBH, and
- Ya and Yb are a BBH.
The basic notion of a PCBBH is really quite intuitive and is shown in Fig. 1.
|
|
Figure 1: The concept of a PCBBH |
It is reasonably straightforward to compute all PCBBHs for a set of genomes, and we have done so for 24 prokaryotic genomes maintained within the WIT system developed at Argonne National Laboratory by R. Overbeek et al. [2, 3]. We employed data from a number of partial genomes as well as complete genomes (hence we altered the definition of BBH to state that there is "no known gene" more similar, rather than "no gene" more similar).
The existence of a pair of close BBHs is certainly a good indication that there might actually be two operons in two genomes, each containing a pair of corresponding orthologs. However, this is not always the case. Our hope is that with enough genomes, the cases in which the implication does not hold will be "washed out with large numbers". It should be noted that the vast majority of PCBBHs derived from closely related organisms (say, two strains of the same species) are of no significance whatsoever and certainly should not be used to infer functional coupling. Conversely, PCBBHs from distantly related organisms are of high significance, since they are unlikely to occur because of chance alone, and can be used to infer functional coupling.
Throughout the examples discussed in the remainder of this work, the reader will note our use of WIT ids for genes and their products. In WIT the ORF identifiers all have the form Rxxddddd, where xx identifies the organism. The organisms corresponding to these two-letter codes are as shown in Tab. 1.
| AA | Aquifex aeolicus | MP | Mycoplasma pneumoniae |
|---|---|---|---|
| AG | Archaeoglobus fulgidus | MT | Mycobacterium tuberculosis |
| BS | Bacillus subtilis | NG | Neisseria gonorrhoea |
| BB | Borrelia burgdorferi | NM | Neisseria meningitidis |
| CA | Clostridium acetobutylicum | PA | Pseudomonas aeruginosa |
| CY | Synechocystis sp. | PF | Pyrococcus furiosus |
| DR | Deinococcus radiodurans | PH | Pyrococcus horikoshii |
| EC | Escherichia coli | PN | Streptococcus pneumoniae |
| HI | Haemophilus influenzae | RC | Rhodobacter capsulatus SB1003 |
| HP | Helicobacter pylori | ST | Streptococcus pyogenes |
| MG | Mycoplasma genitalium | TP | Treponema pallidum |
| MJ | Methanococcus jannaschii | TH | Methanobacterium thermoautotrophicum |
We will use these codes as column headers in tables below.
Given a set of PCBBHs, we can use them to amass evidence that two particular genes on a given genome are actually co-expressed (and are therefore likely to be functionally coupled). The process proceeds as follows:
MG155 (sp|P47401),
MG154 (sp|P47400).
These IDs correspond to two ORFs from the Mycoplasma genitalium genome sequenced by TIGR [4].
This pair of genes occurs in PCBBHs that include Mycoplasma genitalium and each of eighteen other organisms (AG, BS, BB, DR, EC, HI, HP, TH, MJ, MT, MP, NG, NM, PA, ST, PN, CY, and TP). Clearly, this amounts to an overwhelming amount of evidence that the co-occurrences are not random. In fact, this pair of genes correspond to
MG155 (sp|P47401) SSU ribosomal protein S19Pand these functional roles occur within the same operon in many organisms.
MG154 (sp|P47400) LSU ribosomal protein L2P
is far stronger than justP1
P2
Assuming that none of the genomes are extremely close, the existence of a triangular relationship (which was inspired by a similar restriction employed by Koonin et al. in their definition of COGs [5] should therefore be given more weight than a set of binary connections only.P1
We have experimented with several scoring algorithms, and the final results seem to be almost identical. The algorithm used to produce the results presented in this paper was as follows:
Given a pair of genes Xa and Ya from genome Ga, the score reflecting the evidence that they co-occur was computed by adding an increment for each pair (Xi,Yi) from genomes Gi for which (Xa,Ya) and (Xi,Yi) form a PCBBH:
MinD = the minimum ofAdd MinD to the score.
- the distance between Ga and Gb,
- the distance between Ga and Gi, and
- the distance between Gi and Gb.
The result of summing these increments is the score that offers a rough measure that the co-occurrences of Xa and Ya are meaningful.
In forming these scores, we used the distances from the phylogenetic tree distributed by the Ribosomal Database Project [6] as estimates of the "distance between genomes". These distances range from a minimum of 0 (between two strains of Rhodobacter capsulatus) and 1.53 (between Mycoplasma genitalium and Methanobacterium thermoautotrophicum). At an intuitive level, it would seem that a score greater than about 1.0 would certainly indicate that a careful examination of the evidence is warranted. In fact, we believe that any score greater than about 0.1 is quite suggestive and often does indicate a real functional coupling of the genes. Once we have scored all pairs of genes for which PCBBHs exist, the entire list of gene pairs is sorted by score (with higher scores corresponding to pairs of genes Xa and Ya for which functional coupling is more likely). The central question then becomes:
Are the pairs with relatively high scores actually functionally coupled?
Clearly many pairs of genes that are, in fact, functionally coupled will not show up with high scores. However, it will become clear that almost all the pairs with relatively high scores are functionally coupled.
For the 24 genomes we included in the analysis, we computed 34,644 PCBBHs. Using these PCBBHs, we computed scores for 23,144 pairs of genes. Of these, 10,531 pairs had scores greater than 1.0; 17,247 had scores greater than 0.1.
We will argue that scores above 0.1 do, in fact, indicate probable functional coupling, which motivates the following definition:
Genes X and Y from G form a "clustered pair (CP)" if and only if the score assigned to the two genes by the method described above is equal to or greater than 0.1.
Below we will present anecdotal evidence that a clustered pair actually represents a useful clue in determination of gene function. Before we discuss specific examples, however, let us describe an experiment that we used to evaluate the significance of clustered pairs, by examining what fraction of a set of ORFs that we already have good reason to believe are functionally coupled appear in CPs as a function of the score-threshold.
We examined the pathways in the MPW Database [16] and selected those pathways that contain one or more functional roles (normally enzymes) occurring only in that single pathway, and no other. For every organism in WIT that has been asserted to utilize pathways in this set, we then selected those ORFs assigned to each of these "single-pathway functions"; we found 1720 such ORFs.
We then examined how many of these 1720 ORFs were in CPs, as well as how many of those CPs were with other ORFs in the same pathway. Since each of the selected ORFs occurs in only a single pathway, and since two ORFs believed to code for functions in the same pathway are almost certainly functionally coupled, the fraction of pairs containing a selected ORF and an ORF from the same pathway that are also CPs (i. e., that have scores exceeding the chosen threshold) should provide a rough measure of the significance of CPs at that threshold. We found that, of these 1720 selected ORFs:
- 1044 were not a member of any CP (i. e., were not in any pair having a score above the cutoff-threshold using the described technique).
- Of the remaining 676 ORFs:
- 354 ORFs were connected via CPs to another ORF with a functional role from the same pathway; often, more than one link existed for a given ORF. There were 636 CPs linking these 354 ORFs to other functional roles within the same pathway.
- The remaining 322 ORFs were connected via 1122 CPs to ORFs that either have not yet been assigned a functional role or were assigned a functional role that is in a different pathway. Manual analysis of the latter class of CPs showed that the vast majority of cases we have examined also represent either known or biochemically plausible examples of functional coupling.
Thus, out of a total of 1758 CPs (636 connections between ORFs within the same pathway, plus 1122 connections that are not in the same pathway), at least 36% (636/1758) almost certainly represent clear instances of known functional couplings, by virtue of the facts that both members of the CP have been assigned to have functions that are in the same metabolic pathway, and that at least one of them has been assigned a function that occurs in no other pathway.
Note that this estimate of 36% is a conservative lower bound, because ORFs that have not yet been assigned a function have been lumped in with ORFs that have been assigned a function that is in a different pathway. Note also that, just because two ORFs are not in the same metabolic pathway, it does not imply that they might not be functionally coupled, since the MPW pathways are not all independent -- many of them are in fact subsets of metabolic networks. Indeed, our manual analysis suggests that the majority of the remaining CPs also appear to represent what we believe to be real and biochemically justifiable instances of functional couplings -- they simply did not fit into the highly restrictive set of conditions imposed by our tests.
The results of this experiment are summarized in Tab. 2 below.
Table 2: Analysis of the ORFs with a functional roles that
occur in a single pathway using the functional coupling algorithm.
|
|
CP Scores > 0.1 | |
|
ORFs in 24 genomes with a function that occurs in a single pathway only:
1720 |
ORFs with a function that occurs in a single pathway only,
but are not members of a CP:
1044 |
|
|
ORFs with a function that occurs in a single pathway only,
and are members of a CP:
676 |
Number of ORFs connected by a CP to another ORF
that has an assigned function that is in the same pathway:
354 (connected by 636 CPs) |
|
|
Number of ORFs connected by a CP to another ORF
that either has no assigned function,
or has been assigned a function
in a different pathway:
322 (connected by 1122 CPs) |
||
We then repeated the same steps using a cutoff of 1.0 for the calculation of CPs (i.e., we altered the definition of CP). In this case, somewhat more than half as many CPs were detected (957 for a 1.0 cutoff, versus 1758 for a 0.1 cutoff). However, the ratio of the number of connections between genes within the same pathway to the total number of connections rises only slightly, from 36% to 38%. We consider this to be a strong indication that, while a score of 1.0 is certainly far stronger than a score of 0.1, both scores are excellent indicators of functional coupling.
This "experiment" is strongly suggestive that PCBBHs do reflect actual functional coupling. However we have not yet been able to accurately quantify the extent to which such evidence can be relied upon. We believe that the real value of this technology will become truly established only by actual verification of the hundreds of predictions that can easily be made from the existing data.
Tab. 3 offers an extraction of some pairwise scores to give a feel for the output.
Table 3: Pairwise Scores
| Pairwise score | Organism | WIT id | Other names | Assigned Function |
| 35.51 | Archaeoglobus fulgidus | RAG37125 | AF1922 gi|2648624 | LSU ribosomal protein L2P |
| RAG47409 | AF1921
gi|2648642 |
SSU ribosomal protein S19P | ||
| 19.26 | Methanobacterium thermoautotrophicum | RTH01372 | trpA MTH1660
gi|2622788 |
TRYPTOPHAN SYNTHASE ALPHA CHAIN (EC 4.2.1.20) |
| RTH02024 | trpB MTH1659
gi|2622787 |
TRYPTOPHAN SYNTHASE BETA CHAIN (EC 4.2.1.20) | ||
| 18.38 | Archaeoglobus fulgidus | RAG45695 | dppD
gi|2648780 |
DIPEPTIDE TRANSPORT SYSTEM PERMEASE PROTEIN DPPD |
| RAG45696 | dppC
gi|2648779 |
DIPEPTIDE TRANSPORT SYSTEM PERMEASE PROTEIN DPPC | ||
| 13.96 | Methanobacterium thermoautotrophicum | RTH01473 | dnaJ
gi|2622399 |
DNAJ PROTEIN |
| RTH01629 | dnaK
gi|2622398 |
DNAK PROTEIN | ||
| 11.72 | Deinococcus radiodurans | RDR01648 | infA | INITIATION FACTOR IF-1 |
| RDR01651 | rpsK | SSU ribosomal protein S11P | ||
| 11.38 | Treponema pallidum | RTP00100 | ntpI or ntpM | V-TYPE SODIUM ATP SYNTHASE SUBUNIT I (EC 3.6.1.34) |
| RTP00107 | ntpB | V-TYPE SODIUM ATP SYNTHASE SUBUNIT B (EC 3.6.1.34) | ||
| 11.11 | Deinococcus radiodurans | RDR02462 | aroK | SHIKIMATE KINASE (EC 2.7.1.71) |
| RDR02463 | aroB | 3-DEHYDROQUINATE SYNTHASE (EC 4.6.1.3) | ||
| 10.53 | Methanobacterium thermoautotrophicum | RTH00210 | trpE
gi|2622783 |
ANTHRANILATE SYNTHASE COMPONENT I (EC 4.1.3.27) |
| RTH00596 | trpD
gi|2622789 |
ANTHRANILATE PHOSPHORIBOSYLTRANSFERASE (EC 2.4.2.18) | ||
| 8.45 | Helicobacter pylori | RHP00672 | trpB
sp|P56142 |
TRYPTOPHAN SYNTHASE BETA CHAIN (EC 4.2.1.20) |
| RHP00673 | trpC-trpF
gi|2314446 |
INDOLE-3-GLYCEROL PHOSPHATE SYNTHASE (EC 4.1.1.48) / N-(5'-PHOSPHO-RIBOSYL)ANTHRANILATE ISOMERASE(EC 5.3.1.24) | ||
| 5.81 | Clostridium acetobutylicum | RCA00929 | thyBA | THYMIDYLATE SYNTHASE (EC 2.1.1.45) |
| RCA00930 | dfrA | DIHYDROFOLATE REDUCTASE (EC 1.5.1.3) | ||
| 4.45 | Bacillus subtilis | RBS03520 | ftsX gi|2618835 | CELL DIVISION PROTEIN FTSX |
| RBS03521 | ftsE
gi|2618833 |
CELL DIVISION ATP-BINDING PROTEIN FTSE | ||
| 3.11 | Bacillus subtilis | RBS01632 | fliN
sp|P24073 |
FLAGELLAR MOTOR SWITCH PROTEIN FLIN |
| RBS01636 | fliQ
sp|P35535 |
FLAGELLAR BIOSYNTHETIC PROTEIN FLIQ | ||
| 3.00 | Clostridium acetobutylicum | RCA00178 | tpi
gi|2829140 |
TRIOSEPHOSPHATE ISOMERASE (EC 5.3.1.1) |
| RCA00179 | pgmI
gi|2829141 |
2,3-BISPHOSPHOGLYCERATE-INDEPENDENT PHOSPHOGLYCERATE MUTASE (EC 5.4.2.1) | ||
| 1.91 | Escherichia coli | REC06155 | gyrB gi|1790134 | DNA GYRASE SUBUNIT B (EC 5.99.1.3) |
| REC06549 | dnaA
sp|P03004 |
CHROMOSOMAL REPLICATION INITIATOR PROTEIN DNAA | ||
| 1.40 | Escherichia coli | REC00696 | sdhD
sp|P10445 |
SUCCINATE DEHYDROGENASE HYDROPHOBIC MEMBRANE ANCHOR PROTEIN |
| REC00697 | sdhA
sp|P10444 |
SUCCINATE DEHYDROGENASE FLAVOPROTEIN SUBUNIT (EC 1.3.99.1) | ||
| 0.58 | Escherichia coli | REC00555 | ylcD
sp|P77239 |
HYPOTHETICAL 44.3 KD PROTEIN IN NFRB-PHEP INTERGENIC REGION PRECURSOR |
| REC00556 | ybdE
sp|P38054 |
HYPOTHETICAL 114.7 KD PROTEIN IN NFRB-PHEP INTERGENIC REGION | ||
| 0.58 | Clostridium acetobutylicum | RCA00176 | gap gi|2829138 | GLYCERALDEHYDE 3-PHOSPHATE DEHYDROGENASE (EC 1.2.1.12) |
| RCA00178 | tpi
gi|2829140 |
TRIOSEPHOSPHATE ISOMERASE (EC 5.3.1.1) |
It should be noted that of the 2,545 pairs with scores greater than 1.0, 725 included at least one gene that we have not yet been able to assign a reliable function to based on homology or other evidence; inferences about their functional couplings to other genes should provide valuable additional clues to identifying the functions of many of these ORFs.
To offer anecdotal evidence of the ability of these simple computations to effectively infer functional coupling, we looked at several fairly complex known operons and at the coupling scores produced for the genes in these operons.
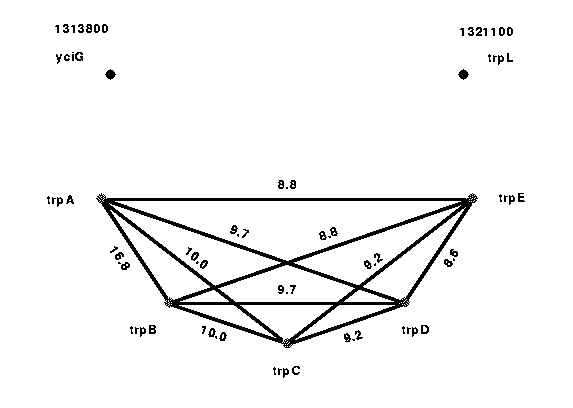
To illustrate an efficiency of the described method in finding functional connections between the genes, we present the results of our analysis of 1313800 -- 1321100 region on the Escherichia coli chromosome (see Fig. 2).
|
Figure 2: Functional coupling between the ORFs in the trp region on the E. coli chromosome. |
This region contains trp operon with the genes trpE, trpD, trpC, trpB and trpA. These genes code for the following enzymes: trpE - anthranilate synthetase (EC 4.1.3.27); trpD - glutamine amidotransferase-phophoribosyl anthranilate transferase; trpC - N-(5-phosphoribosyl)anthranilate isomerase/indole-3-glycerolphosphate synthetase; trpB - tryptophan synthetase (EC 4.2.1.20) beta chain; and trpA - tryptophan synthetase (EC 4.2.1.20) alpha chain. Solid lines connect clustered pairs; the numbers associated with the lines are the pairing scores (see above) which reflect the strength of the connections.
As one can see from Figure , TrpE, TrpD, TrpC, TrpB, and TrpA gene products form a highly conserved PCBBH cluster, which could be viewed as a potential operon. Each ORF in this cluster forms a high-scoring pair with the other members of the cluster, but not with any other ORF in the genome. All five members of this cluster belong to a well-studied tryptophan biosynthesis operon (for review see C. Yanofsky, 1996) [7], which has become the basic reference structure for studies on tryptophan metabolism. As was shown by C. Yanofsky et al. [8], the full-length polycistronic trp mRna encodes the five trp polypeptides (corresponding to trpA, trpB, trpC, trpD, and trpE genes) -- the enzymes of the tryptophan biosynthetic pathway. Our prediction of the potential operon therefore agrees with the literature.
Three different pathways of D,L-diaminopimelate and L-lysine
synthesis are known in prokaryotes [9].
The pathway shown below represents the lysine branch of the aspartic
amino acid family biosynthetic pathway [10].
This pathway is implemented by nine distinct reactions catalyzed by the
enzymes shown in Tab. 4. The PCBBHs relevant to this pathway are shown as well.
Table 4: Functional couplings between the enzymes of the lysine biosynthetic pathway.
| Enzymes, catalysing the consecutive reactions in the pathway | Gene name(s) | Coupled ORFs | ||
|---|---|---|---|---|
| ASPARTATE KINASE (EC 2.7.2.4) | ask; thrA; metL; lysC | |||
| ASPARTATE-SEMIALDEHYDE DEHYDROGENASE (EC 1.2.1.11) | asd | RCY44336 | RPN01355 | RBS01675 |
| DIHYDRODIPICOLINATE SYNTHASE (EC 4.2.1.52) | dapA | RCY38981 | RPN01356 | RBS01677 |
| RCA03322 | RAG28035 | RTH00849 | ||
| DIHYDRODIPICOLINATE REDUCTASE (EC 1.3.1.26) | dapB | RCA03321 | RAG48606 | RTH01270 |
| ACETYL-L,L-DIAMINOPIMELATE AMINOTRANSFERASE | No sequences in the databases | |||
| TETRAHYDRODIPICOLINATE ACETYLTRANSFERASE | No sequences in the databases | |||
| N-ACETYLDIAMINOPIMELATE DEACETYLASE (EC 3.5.1.47) | No sequences in the databases | |||
| DIAMINOPIMELATE EPIMERASE (EC 5.1.1.7) | dapF | RPA02459 | RTH01501 | |
| DIAMINOPIMELATE DECARBOXYLASE (EC 4.1.1.20) | lysA | RPA02462 | RTH01007 | |
Here, we have shown in distinct colors genes from distinct organisms that together form PCBBHs indicating linkages of function. From these PCBBHs, one can infer a tenuous connection between the three functions 1.2.1.11, 4.2.1.52, and 1.3.1.26, as well as the last two functions 4.1.1.20 and 5.1.1.7. Note, however, that no operon in any of the genomes actually couples more than two of these functions. The overall functional coupling emerges much like a holographic image from the set of genomes, but is not present in any single genome. We conjecture that the apparent lesser degree of "completeness" or "maturity" seen in the inferred connections between functions in this pathway might be the result of weaker evolutionary forces pushing these genes toward co-regulation, as compared to those that produced the obvious operons of translation, transcription, and other central cellular mechanisms. In cases such as shown in Tab. 4, we may still gain significant clues leading to meaningful inferences of functional coupling; however, a complete and compelling picture of such pathways might not fully emerge until we have many hundreds of genomes.
We have constructed a formatted version of these pairwise scores and made them available on the WWW via the WIT system [2, 3].
The PCBBHs that connect the enzymes participating in de novo synthesis of IMP are shown in Tab. 5. In this table each row corresponds to a single functional role -- an enzyme catalyzing a particular step in the Purine biosynthetic pathway. Occurrences of these particular functional roles in each genome are presented in each column of the table. We use distinct colors to show potential operon groupings within a column (that is, ORFs sharing a common color within a column are all close on the chromosome in the sense discussed in Definition 1). The first twelve rows of the table correspond to the known enzymatic roles in this pathway. The last row contains evidence that an unknown protein present in seven of the genomes is somehow functionally coupled to IMP biosynthesis.
Table 5: Functional couplings between the enzymes, participating in de novo synthesis of IMP.
| Enzymes of Purine Biosynthetic Pathway | DR | CY | ST | PN | BS | CA | MT | PA | EC | HI | AG | TH | MJ | PF |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AMIDOPHOSPHORIBOSYLTRANSFERASE (EC 2.4.2.14) | - | - | RST01489 | RPN00061 | RBS00650 | RCA01861 | - | - | - | - | - | - | RMJ08546 | - |
| PHOSPHORIBOSYLAMINE-GLYCINE
LIGASE (EC 6.3.4.13) | RDR00668 | - | RST01483 | RPN00065 | RBS00654 | RCA01857 | - | - | REC06280 | RHI05498 | - | - | - | - |
| PHOSPHORIBOSYLGLYCINAMIDE
FORMYLTRANSFERASE (EC 2.1.2.2) | - | - | RST01487 | RPN00063 | RBS00652 | RCA01859 | RMT00037 | - | REC02440 | RHI15219 | - | - | - | - |
| PHOSPHORIBOSYLFORMYLGLYCINAMIDINE
SYNTHASE (EC 6.3.5.3) | RDR03710 | RCY15677 | - | - | RBS00648 | - | RMT00893 | - | - | - | - | RTH01934 | - | RPF00609 |
| PHOSPHORIBOSYLFORMYLGLYCINAMIDINE
SYNTHASE II (EC 6.3.5.3) | RDR03711 | - | RST01490 | RPN00060 | RBS00649 | - | - | - | - | - | RAG50222 | - | - | RPF00610 |
| PHOSPHORIBOSYLFORMYLGLYCINAMIDINE
CYCLO-LIGASE (EC 6.3.3.1) | - | - | RST01488 | RPN00062 | RBS00651 | RCA01860 | - | - | REC02439 | RHI15218 | - | - | RMJ11899 | - |
| PHOSPHORIBOSYLAMINOIMIDAZOLE CARBOXYLASE
ATPASE SUBUNIT (EC 4.1.1.21) | RDR01499 | - | RST01481 | RPN00067 | RBS00644 | - | RMT02837 | RPA03721 | REC04505 | RHI10627 | - | - | - | RPF00920 |
| PHOSPHORIBOSYLAMINOIMIDAZOLE CARBOXYLASE
CATALYTIC SUBUNIT (EC 4.1.1.21) | RDR01497 | - | RST01482 | RPN00066 | RBS00643 | RCA01863 | RMT02836 | RPA03720 | REC04506 | RHI10626 | - | - | - | RPF00919 |
| PHOSPHORIBOSYLAMINOIMIDAZOLE-
SUCCINOCARBOXAMIDE SYNTHASE (EC 6.3.2.6) | - | - | RST01491 | RPN00059 | RBS00646 | RCA01862 | - | - | - | - | - | RTH00832 | RMJ01710 | - |
| ADENYLOSUCCINATE LYASE (EC 4.3.2.2) | - | - | RST01479 | RPN00069 | RBS00645 | - | - | - | - | - | - | - | - | - |
| PHOSPHORIBOSYLAMINOIMIDAZOLECARBOXAMIDE
FORMYLTRANSFERASE (EC 2.1.2.3) / IMP CYCLOHYDROLASE (EC 3.5.4.10) | - | - | RST01486 | RPN00064 | RBS00653 | RCA01858 | RMT00036 | - | REC06281 | RHI09169 | - | - | - | - |
| unknown | - | RCY24689 | - | - | RBS00647 | - | RMT00891 | - | - | - | RAG29290 | RTH01716 | RMJ00185 | RPF00608 |
One notable feature of this example is how the evidence from fourteen distinct genomes combines together to present a vivid portrait of the pathway, as well as the possible coupling of ORFs of unknown function to this pathway. In particular, note that one would still have been able to reconstruct this pathway (as well as infer that it is connected to a protein of unknown function) even if the complete version of it in B. subtilis (BS) and the nearly complete versions in S. pneumoniae (PN) and S. pyogenes (ST) had not been included. Again, one sees that the pathways appear to emerge ``holographicly'' from sets of operons having a greater or lesser degree of ``completeness'' or ``maturity'' in different organisms.
It is known that genes that do not have obvious functional connections
may be co-transcribed [14,15]. Recognition of these atypical operons
could provide very important insights into the interconnections between
the functional subsystems and regulatory mechanisms involved in complex
biological processes.
Our analysis detected a cluster of genes composed of the ribosomal protein
S2P (rpsB), elongation factor Ts (tsf), the ribosome recycling
factor (rrf), the enzyme phosphatidate cytidylyltransferase (cdsA),
uridylate kinase (pyrH), and two hypothetical proteins yaeS
and yaeL.
Using the technique described above, we analyzed functional coupling
between the ORFs in this region in several completely sequenced genomes.
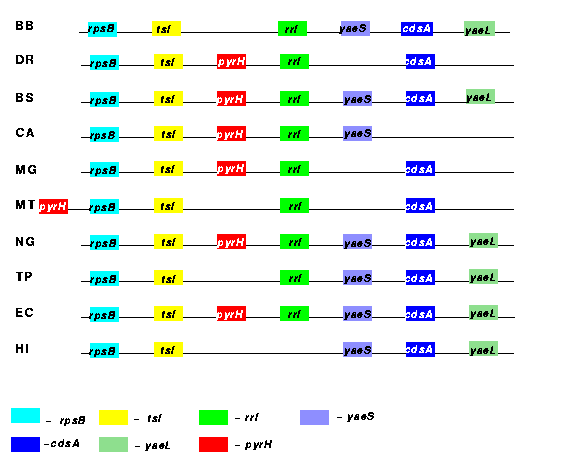
As one can see from Fig. 3, in most of the genomes under consideration rpsB, tsf, pyrH and frr form a highly conserved gene cluster, which includes the uncharacterized genes yaeS and yaeL. With a few exceptions the cluster also contains cdsA, which encodes CDP- diacylglycerol synthase (EC 2.7.7.41).
|
Figure 3: The rps--yae region on the chromosome from ten organisms. |
The order of the genes in the described cluster is preserved in all genomes except M. tuberculosis, in which the pyrH gene precedes rpsB. The functional connections between the genes in this cluster are not obvious. Known experimental data, however, could provide some valuable insights.
It is known that the ribosome recycling factor is an essential protein for bacterial life. The two known functions of ribosome recycling factor (rrf, originally called ribosome releasing factor), are described in [11]: The first function relates to the disassembly of the termination complex, which consists of mRNA, tRNA, and the ribosome bound to the mRNA at the termination codon. The second function of rrf is to prevent errors in translation. In polyphenylalanine synthesis programmed by polyuridylic acid, misincorporation of isoleucine, leucine, or a mixture of amino acids was stimulated up to 17-fold when rrf was omitted from the in vitro system. rrf did not influence the large error (10-fold increase) induced by streptomycin. This means that rrf participates not only in the disassembly of the termination complex but also in peptide elongation.
Yamanaka et al. [12] demonstrated that pyrH gene (formerly smbA) [13], which encodes uridylate kinase (an enzyme participating in pyrimidine biosynthesis), is also involved in chromosome partitioning in E. coli by suppressing mukB. purH was also found to be essential for cell proliferation in the range from 22 to 42 degrees C. Cells that lacked the pyrH protein ceased macromolecular synthesis. The pyrH mutants are sensitive to a detergent, sodium dodecyl sulfate, and they show a novel morphological phenotype under nonpermissive conditions, suggesting a defect in specific membrane sites.
These observations provide only a very indirect grasp of any functional coupling; however, the functions all do appear to be essential during rapid growth of a cell. It may well turn out that the conserved positional relationship of these genes does not convey significant information relating to functional coupling (and there are many instances like this). However, the number of cases in which the functional coupling is obvious suggests that these cases should be considered carefully before dismissing them as accidental curiosities.
The results of our analysis of the potential operons in 24 genomes are available at the following locations:
The ability of such a simple computation to produce such detailed insights into functional coupling is striking. In addition, it must be remembered that our results were produced from only 24 genomes, many of which were incomplete. The ability of the technique proposed here to accurately determine functional coupling will improve dramatically as the number of genomes included in the analysis increases; a crude analysis suggests that the set of PCBBHs should grow as the square of the number of genomes. Given the level of detailed functional data that can be immediately inferred from these PCBBHs, it might well be the case that the cheapest way to acquire detailed evidence of functional coupling would be to rapidly sequence another 50-100 genomes to the point where average contig lengths would be 3-5 Kb. To illustrate this point (albeit quite crudely), we present the following short table, which can provide some insight into the rate of growth in the number of PCBBHs, with sufficient evidence to allow inference of functional coupling:
Table 6: Growth of number of PCBBHs with number of genomes.
| Number of Genomes | Number of PCBBHs with Scores > 0.1 | |
|---|---|---|
| 4 | 998 | |
| 8 | 4859 | |
| 16 | 12570 |
The first set of four genomes that we used were
To arrive at the eight genomes, we added
Finally, to arrive at the sixteen, we added
Here, we counted all PCBBHs for which the simple scoring scheme described above produced scores of 0.1 or better (a weak score, but one suggesting a coupling).
It is difficult to make an accurate estimate of how rapidly the set of reliable PCBBHs will grow, given the number of variables in the genomes used for this tabulation (number of ORFs/genome, phylogenetic distribution, size of contigs, differing levels of operons in different domains, etc.). One would expect the growth to be better than linear.
We have described the computation of PCBBHs and illustrated their use by examples that are probably familiar to the reader. However, it should be stressed that the PCBBHs that we can already compute offer what appears to be substantial evidence coupling hundreds of genes with unknown function to complexes of genes for which the function is known. The detailed analysis of this data will require years, but it is already clear that this data provides a remarkably rich set of clues that should play a role in guiding wet lab verification or rejection of a rapidly emerging set of hypothesis.
This work was supported in part by the U.S. Department of Energy under contract W-31-109-Eng-38.